“Three, that’s the Magic Number
Yes, it is, it’s the Magic Number…”
The above two lines are the opening lyrics of De La Souls hit song from the 1980s “The Magic Number”. The number three also happens to be an important number if you are counting Circulating Tumour Cells (CTC) using the Cell Search kit in metastatic colorectal cancer (mCRC). (See mCRC section, p18 onwards, in the marketing brochure here for more details and also a publication here.) That number directly relates to a patients’ survival prognosis: if a patient has 3 or more CTCs prior to treatment, then that patients’ prognosis will be poorer than for a patient with less than 3 CTCs.
You may be wondering, is the survival probability the same for someone who has 4 versus 100 CTCs? When the platform says 3 how accurate is it? Why did I put “Tumour” in quotation marks? In this blog-post we will briefly explore these questions.
Why the quotation marks around Tumour? Our first port of call will be the brochure (link) mentioned above. The first section of the brochure focusses on the Limitations, Expected Values and Performance Characteristics of the kit. If you take a look at Figure 1 on pVII you will see the distribution of CTC counts across numerous metastatic tumour types, benign disease and healthy volunteers. 10 of the 295 health volunteer samples contained a single CTC. This doesn’t mean the healthy volunteers have cancer but simply highlights that the system may also pick up healthy epithelial cells. Therefore, it may be more appropriate to call these cells Circulating Epithelial Cells rather than Circulating Tumour Cells. Discussions I’ve had with scientists at conferences seem to agree with this, as it appears there is a mix of healthy and cancerous epithelial cells within a sample.
Next, how good is the system at sensing the number of tumour cells when you know approximately how many were in the sample to begin with? The answer to this question can be seen in Table 4 on pVII of the marketing brochure (link). What the table highlights is that as you would expect the recovery is not 100% accurate, there is a modest difference between the expected number and the observed in the samples.
In summary there is a degree of noise in the enumeration process of CTCs. This noise may explain why when scientists have searched for thresholds, they have been quite low. It may be that you need to see 3, 4 or 5 to be sure you actually have one genuine CTC in your sample. So, it could be the thresholds being used are simply related to whether or not there are CTCs there in the sample.
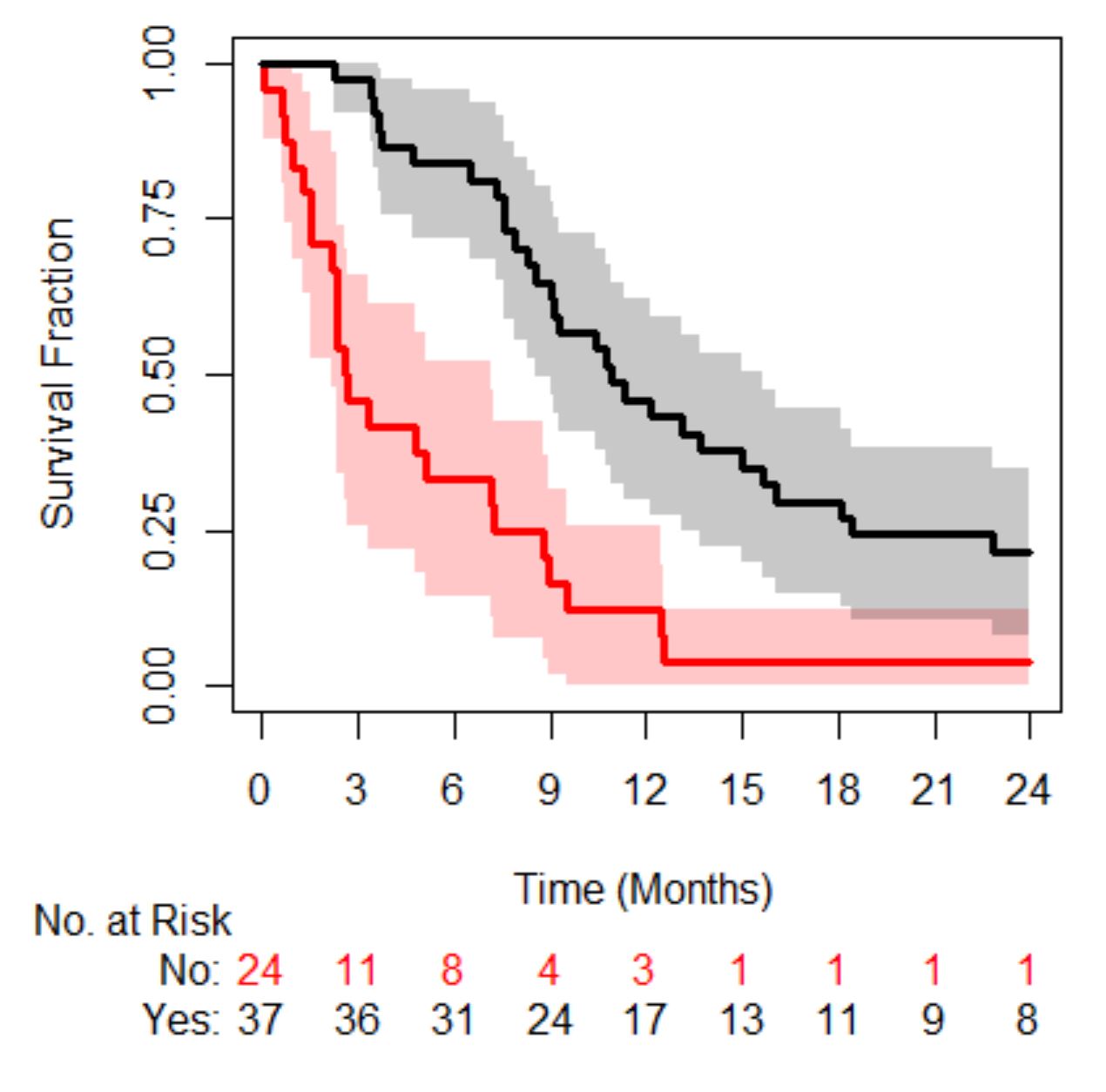
Moving on to the final question, is a patient’s risk of death the same if they have a small number of CTCs versus a large number. In order to answer this, we need a data-set. A digitized data-set from a study in metastatic castrate resistant prostate cancer (mCRPC) will be used, the original study can be read about here and the data-set can be found here with some example code going through the analysis below. The cohort contains 156 patients, 94 deaths with a median survival time of 21 months (95% confidence interval was 16-24 months).
In mCRPC the Magic Number is 5. If patients have less than 5 their prognosis is better than those that have 5 or more. So, the question we are interested in, is the prognosis of a patient with 5 CTCs different from someone who has 100 CTCs?
Below is a figure of the distribution of CTC counts in this cohort of patients. You will notice that there is a large proportion of patients with 0 or 1 values, 38/156 and 15/156 respectively. There is generally quite a wide range of values.

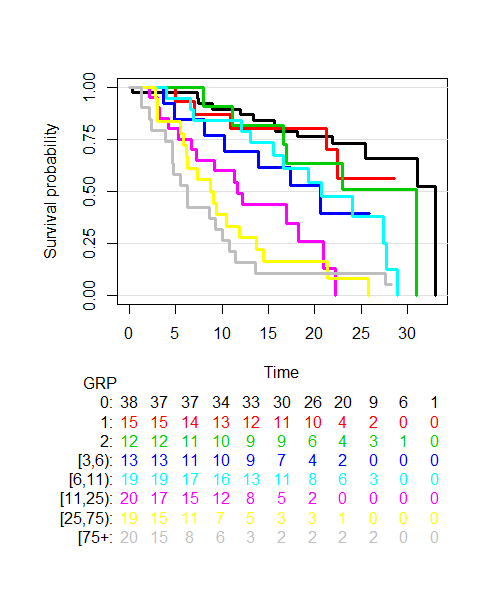
The next plot we shall look at is one of survival probability over time of groups of patients generated by splitting the distribution of CTC counts into 8 groups (generated by looking at the 12.5th, 25th, 37.5th, 50th, 62.5th, 75th and 87.5th percentiles), see below.

The plot shows that as a patient’s CTC count increases their prognosis worsens. Imagine a patient with a CTC count of 5 (located in the dark blue group) versus a patient with CTC count 100 (located in the grey group). It’s clear the prognosis of these two groups is different. Yet if we use the Magic Number 5 both patients will be told they have the same prognosis, which is clearly incorrect. Let’s explore this further and move away from categorising CTC counts…
An alternative way of visualizing the data can be obtained by plotting the log(Hazard Ratio) as a function of CTC counts according to the groups, see plot below.

We can see that the relationship is not quite linear nor does there appear to be an obvious cut-point. In fact, the relationship looks rather sigmoidal, like a Hill function (or to pharmacologists an Emax model). In fact we can fit a Hill function, Hmax/(1+(CTC50/CTC)^h), to the data as shown below. (Hmax, CTC50 and h are parameters that need to be estimated.)

So how does this compare with using the Magic Number 5? In the code, see here, you will see a comparison of model likelihoods which highlights, unsurprisingly, that a sigmoid model better describes the data than using the magic number 5 as well as many other discrimination indexes.
This brief analysis clearly shows using a Magic Number approach to analysing the correlation between CTC counts and prognosis is clearly not in the patient’s favour. Imagine if this was your data, let’s stop dichotomania!