In 11 easy lessons:
Notes:
QEF01 – Introduction to Quantum Economics and Finance
QEF02 – Quantum Probability and Logic
QEF03 – Basics of Quantum Computing
QEF07 – The Prisoner’s Dilemma
QEF09 – Threshold Effects in Quantum Economics

The Minsky model was developed by Steve Keen as a simple macroeconomic model that illustrates some of the insights of Hyman Minsky. The model takes as its starting point Goodwin’s growth cycle model (Goodwin, 1967), which can be expressed as differential equations in the employment rate and the wage share of output.
The equations for Goodwin’s model are determined by assuming simple linear relationships between the variables (see Keen’s paper for details). Changes in real wages are linked to the employment rate via the Phillips curve. Output is assumed to be a linear function of capital stock, investment is equal to profit, and the rate of change of capital stock equals investment minus depreciation.
The equations in employment rate and wage share of output turn out to be none other than the Lotka–Volterra equations which are used in biology to model predator-prey interaction. As employment rises from a low level (like a rabbit population), wages begin to climb (foxes), until wages becomes too high, at which point employment declines, followed by wages, and so on in a repeating limit cycle.
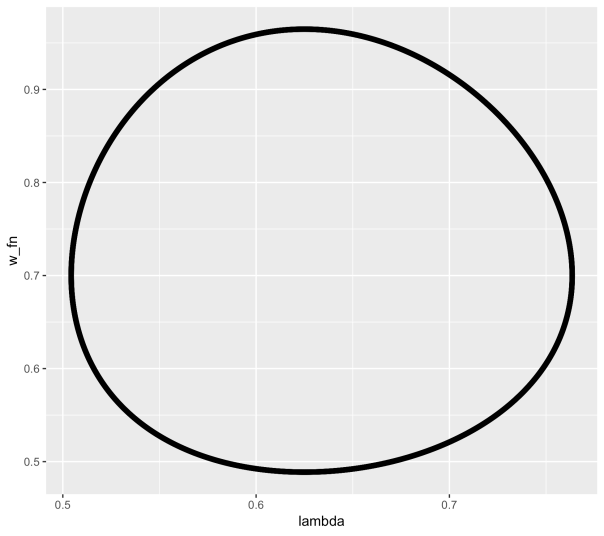
In order to incorporate Minsky’s insights concerning the role of debt finance, a first step is to note that when profit’s share of output is high, firms will borrow to invest. Therefore the assumption that investment equals profit is replaced by a nonlinear function for investment. Similarly the linear Phillips curve is replaced by a more realistic nonlinear relation (in both cases, a generalised exponential curve is used). The equation for profit is modified to include interest payments on the debt. Finally the rate of change of debt is set equal to investment minus profit.
Together, these changes mean that the simple limit cycle behavior of the Goodwin model becomes much more complex, and capable of modelling the kind of nonlinear, debt-fueled behavior that (as Minsky showed) characterises the real economy. A separate equation also accounts for the adjustment of the price level, which converges to a markup over the monetary cost of production.
So how realistic is this model? The idea that employment level and wages are in a simple (linear or nonlinear) kind of predator-prey relation seems problematic, especially given that in recent decades real wages in many countries have hardly budged, regardless of employment level. Similarly the notion of a constant linear “accelerator” relating output to capital stock seems a little simplistic. Of course, as in systems biology, any systems dynamics model of the economy has to make such compromises, because otherwise the model becomes impossible to parameterise. As always, the model is best seen as a patch which captures some aspects of the underlying dynamics.
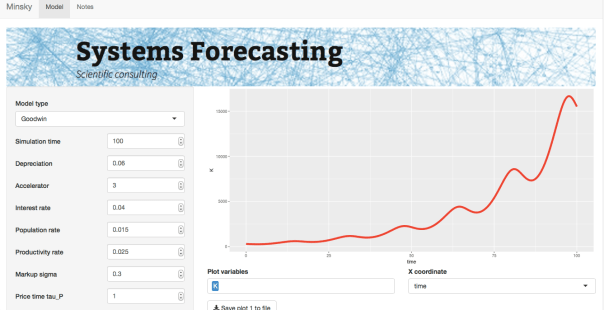
In order to experiment with the model, I coded it up as a Shiny app. The model has rate equations for the following variables: capital K, population N, productivity a, wage rate w, debt D, and price level P. (The model can also be run using Keen’s Minsky software.) Keen also has a version that includes an explicit monetary sector (see reference), which adds a few more equations and more complexity. At that point though I might be tempted to look at simpler models of particular subsets of the economy.
References
Goodwin, Richard, 1967. A growth cycle. In: Feinstein, C.H. (Ed.), Socialism, Capitalism and Economic Growth. Cambridge University Press, Cambridge, 54–58.
Keen, S. (2013). A monetary Minsky model of the Great Moderation and the Great Recession. Journal of Economic Behavior and Organization, 86, 221-235.

Cover article (in Japanese) by David Orrell in Newsweek Japan on why economists can’t predict the future. Read an extract here.
There is growing interest, within the pharmaceutical industry, in using a patients’ own cancer material to screen the effect of numerous treatments within an animal model. The reason for shifting from standard xenografts, based on historical immortalised cell-lines, is that those models are considered to be very different to the patients’ tumours of today. Thus PDX models are considered to be a more relevant model as they are “closer” to the patient population in which you are about to test your new treatment.
Typically only a handful of PDX models are used, but recently there has been a shift in focus to perform population PDX studies which mimic small scale clinical trials. One of these studies by Gao et al. also published, as an excel file in the supplementary information, the raw data which included not only the treatment effect growth curves, but also genomic data consisting of DNA copy number, gene expression and mutation. Using this data it is possible to explore correlations between treatment response and genomic features.
We at Systems Forecasting are always appreciative of freely available data sets, and have designed an equally free and available PDXdata app to browse through this data.
The app can be used to read excel files in the same form as the Novartis file “nm.3954-S2.xlsx”. It translates volume measurements to diameter, and computes a linear fit to each tumour growth time series. The user can then plot time series, organised by ID or by treatment, or examine statistics for the entire data set. The aim is to explore how well linear models can be used to fit this type of data.
The “Diameters” page shown in the figure below is used to plot time series for data. First read in the excel data file; this may take a while, so a progress bar is included. Data can be grouped either by ID(s) or by treatment(s). Note the Novartis data has several treatments for the same ID, and the data is filtered to include only those IDs with an untreated case. If there is only one treatment per ID, one can group by treatment and then plot the data for the IDs with that treatment. In this case the untreated fit is computed from the untreated IDs.

As shown in the next figure, the “Copy Number” and “RNA” tabs allow the user to plot the correlations between copy number or RNA and treatment efficacy, as measured by change in the slope of linear growth, for individual treatments (provided data is available for the selected treatment).

Finally, the “Statistics” page plots a histogram of data derived from the linear models. These include intercept, slope, and sigma for the linear fit to each time series; the difference in slope between the treated and untreated cases (delslope); the growth from initial to final time of the linear fit to the untreated case (lingr); and the difference delgr=diamgr-lingr, which measures diameter loss due to drug.

This app is very much a work-in-progress, and at the moment is primarily a way to browse, view and plot the data. We will add more functionality as it becomes available.

BUY ON AMAZON.COM BUY ON AMAZON.CO.UK
Explore the deadly elegance of finance’s hidden powerhouse
The Money Formula takes you inside the engine room of the global economy to explore the little-understood world of quantitative finance, and show how the future of our economy rests on the backs of this all-but-impenetrable industry. Written not from a post-crisis perspective – but from a preventative point of view – this book traces the development of financial derivatives from bonds to credit default swaps, and shows how mathematical formulas went beyond pricing to expand their use to the point where they dwarfed the real economy. You’ll learn how the deadly allure of their ice-cold beauty has misled generations of economists and investors, and how continued reliance on these formulas can either assist future economic development, or send the global economy into the financial equivalent of a cardiac arrest.
Rather than rehash tales of post-crisis fallout, this book focuses on preventing the next one. By exploring the heart of the shadow economy, you’ll be better prepared to ride the rough waves of finance into the turbulent future.
How do you create a quadrillion dollars out of nothing, blow it away and leave a hole so large that even years of “quantitative easing” can’t fill it – and then go back to doing the same thing? Even amidst global recovery, the financial system still has the potential to seize up at any moment. The Money Formula explores the how and why of financial disaster, what must happen to prevent the next one.

PRAISE FOR THE MONEY FORMULA
“This book has humor, attitude, clarity, science and common sense; it pulls no punches and takes no prisoners.”
Nassim Nicholas Taleb, Scholar and former trader
“There are lots of people who′d prefer you didn′t read this book: financial advisors, pension fund managers, regulators and more than a few politicians. That′s because it makes plain their complicity in a trillion dollar scam that nearly destroyed the global financial system. Insiders Wilmott and Orrell explain how it was done, how to stop it happening again and why those with the power to act are so reluctant to wield it.”
Robert Matthews, Author of Chancing It: The Laws of Chance and How They Can Work for You
“Few contemporary developments are more important and more terrifying than the increasing power of the financial system in the global economy. This book makes it clear that this system is operated either by people who don′t know what they are doing or who are so greed–stricken that they don′t care. Risk is at dangerous levels. Can this be fixed? It can and this book full of healthy skepticism and high expertise shows how.”
Bryan Appleyard, Author and Sunday Times writer
“In a financial world that relies more and more on models that fewer and fewer people understand, this is an essential, deeply insightful as well as entertaining read.”
Joris Luyendijk, Author of Swimming with Sharks: My Journey into the World of the Bankers
“A fresh and lively explanation of modern quantitative finance, its perils and what we might do to protect against a repeat of disasters like 2008–09. This insightful, important and original critique of the financial system is also fun to read.”
Edward O. Thorp, Author of A Man for All Markets and New York Times bestseller Beat the Dealer
The Winter 2017 edition of Foresight magazine includes my commentary on the article Changing the Paradigm for Business Forecasting by Michael Gilliland from SAS. Both are behind a paywall (though a longer version of Michael’s argument can be read on his SAS blog), but here is a brief summary.
According to Gilliland, business forecasting is currently dominated by an “offensive” paradigm, which is “characterized by a focus on models, methods, and organizational processes that seek to extract every last fraction of accuracy from our forecasts. More is thought to be better—more data, bigger computers, more complex models—and more elaborate collaborative processes.”
He argues that our “love affair with complexity” can lead to extra effort and cost, while actually reducing forecast accuracy. And while managers have often been seduced by the idea that “big data was going to solve all our forecasting problems”, research shows that even with complex models, forecast accuracy often fails to beat even a no-change forecasting model. His article therefore advocates a paradigm shift towards “defensive” forecasting, which focuses on simplifying the forecasting process, eliminating bad practices, and adding value.
My comment on this (in about 1200 words) is … I agree. But I would argue that the problem is less big data, or even complexity, than big theory.
Our current modelling paradigm is fundamentally reductionist – the idea is to reduce a system to its parts, figure out the laws that govern their interactions, build a giant simulation of the whole thing, and solve. The resulting models are highly complex, and their flexibility makes them good at fitting past data, but they tend to be unstable (or stable in the wrong way) and are poor at making predictions.
If however we recognise that complex systems have emergent properties that resist a reductionist approach, it makes more sense to build models that only attempt to capture some aspect of the system behaviour, instead of reproducing the whole thing.
An example of this approach, discussed earlier on this blog, relates to the question of predicting heart toxicity for new drug compounds, based on ion channel readings. One way to predict heart toxicity based on these test results is to employ teams of researchers to build an incredibly complicated mechanistic model of the heart, consisting of hundreds of differential equations, and use the ion channel inputs as inputs. Or you can use a machine learning model. Or, most complicated, you can combine these in a multi-model approach. However Hitesh Mistry found that a simple model, which simply adds or subtracts the ion channel readings – the only parameters are +1 and -1 – performs just as well as the multi-model approach using three large-scale models plus a machine learning model (see Complexity v Simplicity, the winner is?).
Now, to obtain the simple model Mistry used some fairly sophisticated data analysis tools. But what counts is not the complexity of the methods, but the complexity of the final model. And in general, complexity-based models are often simpler than their reductionist counterparts.
I therefore strongly agree with Michael Gilliland that a “defensive” approach makes sense. But I think the paradigm shift he describes is part of, or related to, a move away from reductionist models, which we are realising don’t work very well for complex systems. With this new paradigm, models will be simpler, but they can also draw on a range of techniques that have developed for the analysis of complex systems.
Back in the early 2000s, I worked a couple of years as a senior scientist at the Institute for Systems Biology in Seattle. So it was nice to revisit the area for the recent Seventh American Conference on Pharmacometrics (ACoP7).
A lot has changed in Seattle in the last 15 years. The area around South Lake Union, near where I lived, has been turned into a major hub for biotechnology and the life sciences. Amazon is constructing a new campus featuring giant ‘biospheres’ which look like nothing I have ever seen.
Attending the conference, though, was like a blast from the past – because unlike the models used by architects to design their space-age buildings, the models used in pharmacology have barely moved on.
While there were many interesting and informative presentations and posters, most of these involved relatively simple models based on ordinary differential equations, very similar to the ones we were developing at the ISB years ago. The emphasis at the conference was on using models to graphically present relationships, such as the interaction between drugs when used in combination, and compute optimal doses. There was very little about more modern techniques such as machine learning or data analysis.
There was also little interest in producing models that are truly predictive. Many models were said to be predictive, but this just meant that they could reproduce some kind of known behaviour once the parameters were tweaked. A session on model complexity did not discuss the fact, for example, that complex models are often less predictive than simple models (a recurrent theme in this blog, see for example Complexity v Simplicity, the winner is?). Problems such as overfitting were also not discussed. The focus seemed to be on models that are descriptive of a system, rather than on forecasting techniques.
The reason for this appears to come down to institutional effects. For example, models that look familiar are more acceptable. Also, not everyone has the skills or incentives to question claims of predictability or accuracy, and there is a general acceptance that complex models are the way forward. This was shown by a presentation from an FDA regulator, which concentrated on models being seen as gold-standard rather than accurate (see our post on model misuse in cardiac models).
Pharmacometrics is clearly a very conservative area. However this conservatism means only that change is delayed, not that it won’t happen; and when it does happen it will probably be quick. The area of personalized medicine, for example, will only work if models can actually make reliable predictions.
As with Seattle, the skyline may change dramatically in a very short time.
The standard way of answering this question is to ask whether the effect could reasonably have happened by chance (the null hypothesis). If not, then the result is announced to be ‘significant’. The usual threshold for significance is that there is only a 5 percent chance of the results happening due to purely random effects.
This sounds sensible, and has the advantage of being easy to compute. Which is perhaps why statistical significance has been adopted as the default test in most fields of science. However, there is something a little confusing about the approach; because it asks whether adopting the opposite of the theory – the null hypothesis – would mean that the data is unlikely to be true. But what we want to know is whether a theory is true. And that isn’t the same thing.
As just one example, suppose we have lots of data and after extensive testing of various theories we discover one that passes the 5 percent significance test. Is it really 95 percent likely to be true? Not necessarily – because if we are trying out lots of ideas, then it is likely that we will find one that matches purely by chance.
While there are ways of working around this within the framework of standard statistics, the problem usually gets glossed over in the vast majority of textbooks and articles. So for example it is typical to say that a result is ‘significant’ without any discussion of whether it is plausible in a more general sense (see our post on model misuse in cardiac modeling).
The effect is magnified by publication bias – try out multiple theories, find one that works, and publish. Which might explain why, according to a number of studies (see for example here and here), much scientific work proves impossible to replicate – a situation which scientist Robert Matthews calls a ‘scandal of stunning proportions’ (see his book Chancing It: The laws of chance – and what they mean for you).
The way of Bayes
An alternative approach is provided by Bayesian statistics. Instead of starting with the assumption that data is random and making weird significance tests on null hypotheses, it just tries to estimate the probability that a model is right (i.e. the thing we want to know) given the complete context. But it is harder to calculate for two reasons.
One is that, because it treats new data as updating our confidence in a theory, it also requires we have some prior estimate of that confidence, which of course may be hard to quantify – though the problem goes away as more data becomes available. (To see how the prior can affect the results, see the BayesianOpionionator web app.) Another problem is that the approach does not treat the theory as fixed, which means that we may have to evaluate probabilities over whole families of theories, or at least a range of parameter values. However this is less of an issue today since the simulations can be performed automatically using fast computers and specialised software.
Perhaps the biggest impediment, though, is that when results are passed through the Bayesian filter, they often just don’t seem all that significant. But while that may be bad for publications, and media stories, it is surely good for science.
A common critique of biologists, and scientists in general, concerns their occasionally overenthusiastic tendency to find patterns in nature – especially when the pattern is a straight line. It is certainly notable how, confronted with a cloud of noisy data, scientists often manage to draw a straight line through it and announce that the result is “statistically significant”.
Straight lines have many pleasing properties, both in architecture and in science. If a time series follows a straight line, for example, it is pretty easy to forecast how it should evolve in the near future – just assume that the line continues (note: doesn’t always work).
However this fondness for straightness doesn’t always hold; indeed there are cases where scientists prefer to opt for a more complicated solution. An example is the modelling of tumour growth in cancer biology.
Tumour growth is caused by the proliferation of dividing cells. For example if cells have a cell cycle length td, then the total number of cells will double every td hours, which according to theory should result in exponential growth. In the 1950s (see Collins et al., 1956) it was therefore decided that the growth rate could be measured using the cell doubling time.
In practice, however, it is found that tumours grow more slowly as time goes on, so this exponential curve needed to be modified. One variant is the Gompertz curve, which was originally derived as a model for human lifespans by the British actuary Benjamin Gompertz in 1825, but was adapted for modelling tumour growth in the 1960s (Laird, 1964). This curve gives a tapered growth rate, at the expense of extra parameters, and has remained highly popular as a means of modelling a variety of tumour types.
However, it has often been observed empirically that tumour diameters, as opposed to volumes, appear to grow in a roughly linear fashion. Indeed, this has been known since at least the 1930s. As Mayneord wrote in 1932: “The rather surprising fact emerges that the increase in long diameter of the implanted tumour follows a linear law.” Furthermore, he noted, there was “a simple explanation of the approximate linearity in terms of the structure of the sarcoma. On cutting open the tumour it is often apparent that not the whole of the mass is in a state of active growth, but only a thin capsule (sometimes not more than 1 cm thick) enclosing the necrotic centre of the tumour.”
Because only this outer layer contains dividing cells, the rate of increase for the volume depends on the doubling time multiplied by the volume of the outer layer. If the thickness of the growing layer is small compared to the total tumour radius, then it is easily seen that the radius grows at a constant rate which is equal to the doubling time multiplied by the thickness of the growing layer. The result is a linear growth in radius. This translates to cubic growth in volume, which of course grows more slowly than an exponential curve at longer times – just as the data suggests.
In other words, rather than use a modified exponential curve to fit volume growth, it may be better to use a linear equation to model diameter. This idea that tumour growth is driven by an outer layer of proliferating cells, surrounding a quiescent or necrotic core, has been featured in a number of mathematical models (see e.g. Checkley et al., 2015, and our own CellCycler model). The linear growth law can also be used to analyse tumour data, as in the draft paper: “Analysing within and between patient patient tumour heterogenity via imaging: Vemurafenib, Dabrafenib and Trametinib.” The linear growth equation will of course not be a perfect fit for the growth of all tumours (no simple model is), but it is based on a consistent and empirically verified model of tumour growth, and can be easily parameterised and fit to data.
So why hasn’t this linear growth law caught on more widely? The reason is that what scientists see in data often depends on their mental model of what is going on.
I first encountered this phenomenon in the late 1990s when doing my D.Phil. in the prediction of nonlinear systems, with applications to weather forecasting. The dominant theory at the time said that forecast error was due to sensitivity to initial condition, aka the butterfly effect. As I described in The Future of Everything, researchers insisted that forecast errors showed the exponential growth characteristic of chaos, even though plots showed they clearly grew with slightly negative curvature, which was characteristic of model error.
A similar effect in cancer biology has again changed the way scientists interpret data. Sometimes, a straight line really is the best solution.
Collins, V. P., Loeffler, R. K. & Tivey, H. Observations on growth rates of human tumors. The American journal of roentgenology, radium therapy, and nuclear medicine 76, 988-1000 (1956).
Laird A. K. Dynamics of tumor growth. Br J of Cancer 18 (3): 490–502 (1964).
W. V. Mayneord. On a Law of Growth of Jensen’s Rat Sarcoma. Am J Cancer 16, 841-846 (1932).
Stephen Checkley, Linda MacCallum, James Yates, Paul Jasper, Haobin Luo, John Tolsma, Claus Bendtsen. Bridging the gap between in vitro and in vivo: Dose and schedule predictions for the ATR inhibitor AZD6738. Scientific Reports, 5(3)13545 (2015).
Yorke, E. D., Fuks, Z., Norton, L., Whitmore, W. & Ling, C. C. Modeling the Development of Metastases from Primary and Locally Recurrent Tumors: Comparison with a Clinical Data Base for Prostatic Cancer. Cancer Research 53, 2987-2993 (1993).
Hitesh Mistry, David Orrell, and Raluca Eftimie. Analysing within and between patient patient tumour heterogenity via imaging: Vemurafenib, Dabrafenib and Trametinib. (Working paper)
Tumour modelling has been an active field of research for some decades, and a number of approaches have been taken, ranging from simple models of an idealised spherical tumour, to highly complex models which attempt to account for everything from cellular chemistry to mechanical stresses. Some models use ordinary differential equations, while others use an agent-based approach to track individual cells.
A disadvantage of the more complex models is that they involve a large number of parameters, which can only be roughly estimated from available data. If the aim is to predict, rather than to describe, then this leads to the problem of overfitting: the model is very flexible and can be tuned to fit available data, but is less useful for predicting for example the effect of a new drug.
Indeed, there is a rarely acknowledged tension in mathematical modelling between realism, in the sense of including lots of apparently relevant features, and predictive accuracy. When it comes to the latter, simple models often out-perform complex models. Yet in most areas there is a strong tendency for researchers to develop increasingly intricate models. The reason appears to have less to do with science, than with institutional effects. As one survey of business models notes (and these points would apply equally to cancer modelling) complex models are preferred in large part because: “(1) researchers are rewarded for publishing in highly ranked journals, which favor complexity; (2) forecasters can use complex methods to provide forecasts that support decision-makers’ plans; and (3) forecasters’ clients may be reassured by incomprehensibility.”
Being immune to all such pressures (this is just a blog post after all!) we decided to develop the CellCycler – a parsimonius “toy” model of a cancer tumour that attempts to capture the basic growth and drug-response dynamics using only a minimal number of parameters and assumptions. The model uses circa 100 ordinary differential equations (ODEs) to simulate cells as they pass through the phases of the cell cycle; however the equations are simple and the model only uses parameters that can be observed or reasonably well approximated. It is available online as a Shiny app.

The CellCycler model divides the cell cycle into a number of discrete compartments, and is therefore similar in spirit to other models that for example treat each phase G1, S, G2, and mitosis as a separate compartment, with damaged cells being shunted to their own compartment (see for example the model by Checkley et al. here). Each compartment has its own set of ordinary differential equations which govern how its volume changes with time due to growth, apoptosis, or damage from drugs. There are additional compartments for damaged cells, which may be repaired or lost to apoptosis. Drugs are simulated using standard PK models, along with a simple description of phase-dependent drug action on cells. For the tumour growth, we use a linear model, based like the Checkley et al. paper on the assumption of a thin growing layer (see also our post on The exponential growth effect).
Dividing the cell cycle into separate compartments has an interesting and useful side effect, which is that it introduces a degree of uncertainty into the calculation. For example, if a drug causes damage and delays progress in a particular phase, then that drug will tend to synchronize the cell population in that state. However there is an obvious difference between cells that are affected when they are at the start of the phase, and those that are already near the end of the phase. If the compartments are too large, that precise information about the state of cells is lost.
The only way to restore precision would be to use a very large number of compartments. But in reality, individual cells will not all have exactly the same doubling time. We therefore want to have a degree of uncertainty. And this can be controlled by adjusting the number of compartments.
This effect is illustrated by the figure below, which shows how a perturbation at time zero in one compartment tends to blur out over time, for models with 25, 50, and 100 compartments, and a doubling time of 24 hours. In each case a perturbation is made to compartment 1 at the beginning of the cell cycle (the magnitude is scaled to the number of compartments so the total size of the perturbation is the same in terms of total volume). For the case with 50 compartments, the curve after one 24 hours is closely approximated by a normal distribution with standard deviation of 3.4 hours or about 14 percent. In general, the standard deviation can be shown to be approximately equal to the doubling time divided by the square root of N.
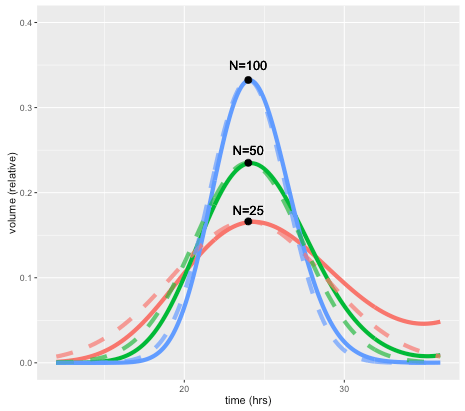
A unique feature of the CellCycler is that it exploits this property as a way of adjusting the variability of doubling time in the cell population. The model can therefore provide a first-order approximation to the more complex heterogeneity that can be simulated using agent-based models. While we don’t usually have exact data on the spread of doubling times in the growing layer, the default level of 50 compartments gives what appears to be a reasonable degree of spread (about 14 percent). Using 25 compartments gives 20 percent, while using 100 compartments decreases this to 10 percent.
The starting point for the Shiny web application is the Cells page, which is used to model the dynamics of a growing cell population. The key parameters are the average cell doubling time, and the fraction spent in each phase. The number of model compartments can be adjusted in the Advanced page: note that, along with doubling time spread, the choice also affects both the simulation time (more compartments is slower), and the discretisation of the cell cycle. For example with 50 compartments the proportional phase times will be rounded off to the nearest 1/50=0.02.
The next pages, PK1 and PK2, are used to parameterise the PK models and drug effects. The program has a choice of standard PK models, with adjustable parameters such as Dose/Volume. In addition the phase of action (choices are G1, S, G2, M, or all), and rates for death, damage, and repair can be adjusted. Finally, the Tumor page (shown below) uses the model simulation to generate a plot of tumor radius, given an initial radius and growing layer. Plots can be overlaid with experimental data.

We hope the CellCycler can be a useful tool for research or for exploring the dynamics of tumour growth. As mentioned above it is only a “toy” model of a tumour. However, all our models of complex organic systems – be they of a tumor, the economy, or the global climate system – are toys compared to the real things. And of course there is nothing to stop users from extending the model to incorporate additional effects. Though whether this will lead to improved predictive accuracy is another question.
Try the CellCycler web app here.
Stephen Checkley, Linda MacCallum, James Yates, Paul Jasper, Haobin Luo, John Tolsma, Claus Bendtsen. “Bridging the gap between in vitro and in vivo: Dose and schedule predictions for the ATR inhibitor AZD6738,” Scientific Reports.2015;5(3)13545.
Green, Kesten C. & Armstrong, J. Scott, 2015. “Simple versus complex forecasting: The evidence,” Journal of Business Research, Elsevier, vol. 68(8), pages 1678-1685.